
新一代组学数据分析平台,实现自然语言和人工
 在过去的二十年里,高通量测序技术分子彻底改变了生物医学的研究模式。从数千名患者、动物模型和细胞系中产生的各种组学数据,如基因组、转录组、蛋白质组、表观基因组和代谢数据等,正在以越来越快的速度积累。
在过去的二十年里,高通量测序技术分子彻底改变了生物医学的研究模式。从数千名患者、动物模型和细胞系中产生的各种组学数据,如基因组、转录组、蛋白质组、表观基因组和代谢数据等,正在以越来越快的速度积累。
这些丰富的组学数据为系统地描述分子机制和开发相关生物医学应用提供了前所未有的资源信息。同时,数据的激增也给数据分析人员提出了一个重大挑战:如何通过海量的数据获得有意义的信息?
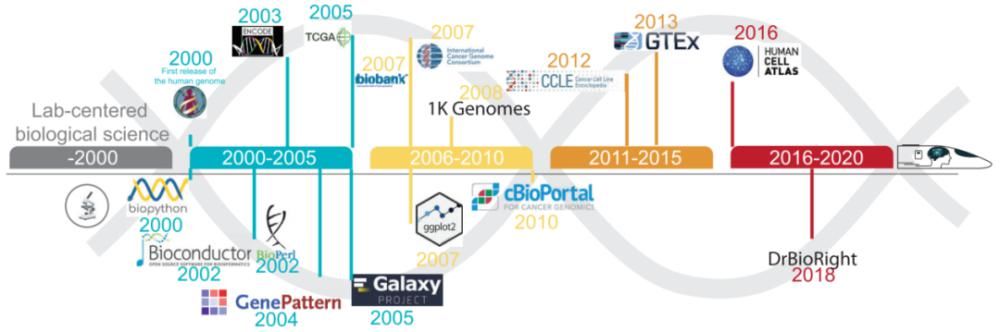 图1.组学数据资源和生物信息学工具的发展。
图1.组学数据资源和生物信息学工具的发展。
近年来,相关领域的人员在克服“解读高通量数据”方面取得了重大进展(如图1)。最初,组学数据通常由生物信息学家或计算生物学家使用通用编程语言(Python、R和Perl)编写的内部脚本进行数据分析。后来,研究人员开发了一些专门的生物信息编程模块的集合,如Biopython、BioPerl、Bioconductor和ggplot,这些模块的推广使组学数据的分析和可视化更加容易。
尽管如此,这些工具仍然需要用户具有一定的编程专业知识,但这是许多实验研究人员所不具备的,因此这严重阻碍了大多数研究人员(特别是那些没有生物信息学和统计专业知识的研究人员)直接充分利用组学数据。因此,开发用户友好的、简单易行的组学数据分析工具就显得尤为重要。
图2.文章发表在Cancer Cell
近日,德克萨斯大学MD安德森癌症中心梁晗教授课题组在Cancer Cell发文报道了一种简单易行的下一代组学数据分析工具---DrBioRight。该工具由研究团队历时三年推出,是一个基于自然语言的人工智能驱动的组学数据分析平台,通过简单的对话就可以进行组学数据的分析和探索。此外,文章详细分析了组学时代数据分析面临的挑战和可行的解决方案。
据文章报道,DrBioRight由两个子系统组成:一个用户友好的网页界面,用以输入研究人员的需求;另外一个是后端计算服务器,用以储存数据和计算分析数据。
与其他生物信息学工具相比,DrBioRight采用了一个简单的在线聊天界面,只有一个输入区和一个输出区,与用户的所有交互都基于人类自然语言,入门简单,无需任何编程语言基础。
例如,用户可以打字输入?“对乳腺癌TP53基因表达进行生存分析”(图3B),以检验TP53基因表达水平与乳腺癌患者总生存是否存在相关性。在接收到输入文本后,DrBioRight将运行它的自然语言处理模块来标记输入的问题,基于在输入中识别的特性,后端AI模块将计算分数来预测最匹配的分析任务,然后程序将调用特定的分析模块,识别相关数据集。
另外,在提交计算任务之前,DrBioRight会询问用户系统检测试别到的任务是否确实是预期的分析。如果确认,作业调度器将把任务提交到作业队列,并使用基于云的计算节点进行处理。一旦任务完成,DrBioRight将调用适当的可视化模块,并将结果发送到输出区域的用户(图3C)。
最后,每次成功执行作业之后,DrBioRight将要求用户进行评价,收集到的反馈将用于进一步提高自然语言处理模块和AI模块的性能。
 图3. DrBioRight分析流程概述。
图3. DrBioRight分析流程概述。
目前,DrBioRight已经策划和加载了一些广泛使用的癌症组学数据集,包括TCGA、国际癌症基因组联盟(ICGC)和癌细胞系百科全书(Cancer Cell Line Encyclopedia),总计已超过20,000个样本的多组学数据。
随着DrBioRight的成功发布,以及在展示了其能力和效用之后,研究人员提出了下一代数据分析应该具备的五个关键特性:
自然语言可及性,即通过日常通用语言就可以达到数据分析的目的;
人工智能性,高通量数据的分析和解读应该逐步由人员驱动向数据驱动转换,即以数据解读数据;
透明性,可重复性是生物医学研究中最看重的一点,因此高通量的数据分析也应该是有据可循,且数据分析细解需公开透明;
移动和社交平台友好性,智能手机作为最便捷的通讯工具,为研究者提供了一个优秀的平台,可以不受地点和时间的限制进行组学数据分析;另外启用社交媒体功能将大大增强高通量数据分析的结果惠及普通大众。
开放性,各领域专家,包括算法开发者,数据科学家,生物学家和临床医生等,需要高效合作,因此,一个开放性的开发中心就显得很必要。
上述这些特性将使生物医学研究社区能够以一种直观、高效、可靠和协作的方式探索多组学数据。
上一篇:石鲁极具个性的笔墨语言对当代山水产生了巨大
下一篇:没有了




